
The poster of Psi-k funded workshops in 2023/2024 is now available to download… https://psi-k.net/workshops/.
Please share to your mailing lists – or print a copy and display it on your noticeboards.

The poster of Psi-k funded workshops in 2023/2024 is now available to download… https://psi-k.net/workshops/.
Please share to your mailing lists – or print a copy and display it on your noticeboards.


Many congratulations to the former Psi-k Chair, and current Trustee, Nicola Marzari and his team on winning the inaugural PRACE HPC Excellence Award for their work in the discovery and characterization of novel two-dimensional materials!


Interested in promoting discussion and fostering collaborations in research areas of broad mutual interest for CECAM and Psi-k?
Submit your expression of interest to organize a CECAM Psi-k Research Conference in 2023 by filling the simple form at:
https://www.cecam.org/submit-psi-k-proposal
Deadline for submission of the expression of interest: 4 September 2022
Diversity (specific expertise in simulation and modelling, geographical, gender, career stage…) and interdisciplinarity among organisers and participants are key evaluation criteria.
The CECAM Psi-k Research Conference is a forum to explore and foster progress in exciting new topics and open questions of interest to different communities in simulation and modeling, rather than another opportunity to showcase consolidated research. The proposed duration and format of the event should reflect this spirit, with ample time set aside for common discussions and informal exchanges.
Details of previous CECAM Psi-k Research Conferences can be found online.
If selected for further evaluation, you will be invited to submit a full proposal – format similar to previous years – at the end of September 2022, with a deadline for submission on 31 October 2022.

Psi-k is a worldwide network of researchers working on the advancement of first-principles computational materials science. Its mission is to develop fundamental theory, algorithms, and computer codes in order to understand, predict, and design materials properties and functions. Key activities of Psi-k are the organization of conferences, workshops, tutorials and training schools as well as the dissemination of scientific thinking in society [Excerpt from the mission statement at http://psi-k.net/ ].
Psi-k typically funds schools (1-2 weeks), workshops (2-3 days), international conferences (2-5 days), and codes/methods tutorials (3 days-1 week), with a focus on electronic-structure methods, developments, and applications. Funding is of the order of 4,000/8,000/12,000 €, depending on size and duration (as a guideline, 30 €/expected participant/day); Psi-k funds around 25-35 activities for every call (see here http://psi-k.net/workshops/ for the 2022-23 activities).
For this coming round, we are particularly keen to support activities that benefit PhD students and early career researchers, who have missed out on in-person events during the pandemic – schools and tutorials in particular. We also continue to encourage the ingenious development of remote delivery methods where appropriate, to widen the benefit of our activities to groups where the cost of in-person attendance may be a barrier.
We have a two-step application process to simplify and streamline applications, elicit more original proposals, improve planning, and avoid duplication of efforts. The present call for outline proposals is for events that will take place between 1 April 2023 and 31 March 2024.
Outline proposals should be submitted online by Monday 18 July 2022 (midnight UK time), describing the planned event (download a draft of the form to see a preview of the questions that you will be asked here). Psi-k Working Groups (http://psi-k.net/groups ) and Trustees (http://psi-k.net/admin/) will either approve this outline proposal for full submission, reject it, or suggest a merger between different activities – you are very welcome to contact the relevant Working Group leaders or members beforehand.
Feedback will be provided the week of Monday 19 September 2022. Outline proposals that have been approved or mergers that have been successfully negotiated will then be invited to submit a full proposal by Monday 17 October 2022. These proposals will then be evaluated and approved – with full or partial funding – or declined at the meeting of the Trustees, Scientific Advisory Committee (http://psi-k.net/scientific-advisory-committee/) and Working Group leaders on Friday 25 November 2022.
The evaluation of the outline proposals and full proposals will be based on
NOTES
Dear Psi-k,
Following on from our email last month, we are excited to share some news about our whole-community Psi-k 2022 Conference in August.
Now that Switzerland has relaxed all restrictions on travel and events, and with the situation generally improving, the Board of Trustees has decided to proceed with a fully in-person event as planned for 22-25 August 2022.
We intend to open the registration system for discounted early-bird rates in the second half of March.
With 7 plenary talks and 42 symposia accommodating 130 invited talks and open slots for over 180 contributed talks, we are very much looking forward to this opportunity to meet in person to exchange scientific ideas, to make new connections and to renew old ones. In particular we believe that this will be important for early career researchers and PhD students, for whom the pandemic has been very disruptive.
All information is on the conference web site: https://www.psik2022.net
Warmest wishes,
Nicola Marzari, Psi-k 2022 Conference Chair
Peter Haynes, Chair of Trustees
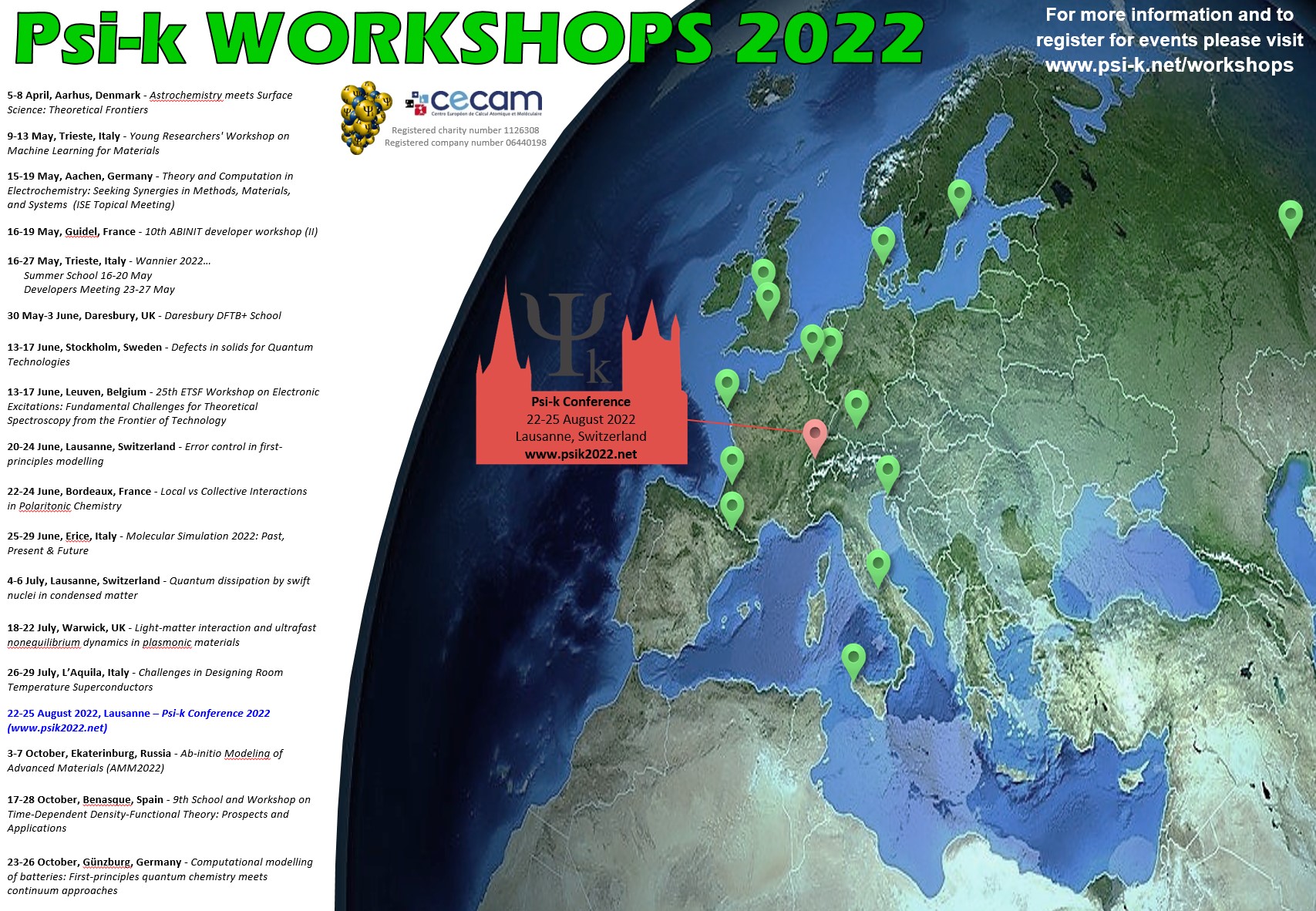
The Psi-k Workshops 2022 Poster is now available to download.
Please help us share information about the 2022 Psi-k funded workshops, schools and training courses by downloading and printing a copy of our poster to display at your institution and sharing with your communities via individual mailing lists.
If you have a twitter account you can also retweet the poster from here.
Thank you for your support.
Dear Psi-k
We hope that you have had a good start to 2022 and are looking forward to more in-person scientific interactions this year.
In particular we have published our programme of workshops on our web site: https://psi-k.net/workshops/

The highlight is of course our whole-community Psi-k Conference in August. We are very encouraged by the positive feedback from the community meeting in November and our Scientific Advisory Committee and are continuing with prudent plans to hold this Conference in person in Lausanne: 22-25 August. We will of course monitor the COVID-19 situation internationally, but at the moment the outlook appears promising in spite of the omicron variant. In particular, meeting in person will be important for the PhD students and early-career researchers in our community.
Please do keep a look out for further announcements about registration – information can be found on the conference web site: https://www.psik2022.net/
Best wishes,
Peter Haynes
Chair of Psi-k Trustees
Nicola Marzari
Chair of Psi-k Conference 2022
